仿製藥製劑出口在當下已成為國內藥企的重要意識,越來越多的製藥企業開始將業務拓展到海外。一方麵海外業務為國內藥企帶來可觀收入,另一方麵被美國或歐盟批準的製劑產品使藥企拿到綠色通行證,獲得更快的國內報批資格。那麽製劑出口需要建立怎樣的體係?研發及生產流程是怎樣的模式?
以美國市場為例,仿製藥製劑產品需要在FDA(美國藥監局)認證的cGMP(動態藥品生產管理規範)條件下生產,並具有與原研產品較高的體內外一致性,才有資格獲批進入美國市場銷售。也就是說,首先,藥企需要擁有一個FDA認證過的cGMP車間;其次是關注產品質量,開展體內外一致性評價研究;最終目標是獲得與原研產品體內生物利用度一致的仿製藥產品,也就是我們常說的通過BE(生物等效性)實驗。
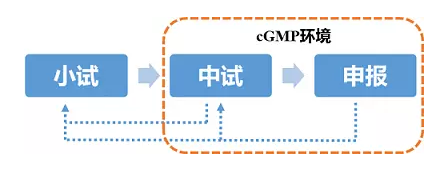
產品的研發從實驗室小試,到車間中試放大,再到申報生產,是一個較為漫長且需經過不斷反複循環的過程,通常經曆3-5年時間。
實驗室小試階段
批量通常在200g~2kg
當仿製藥產品立項後,需要對原研產品進行較為全麵的調研及檢測工作。其目的在於明確研發目標,使研發的每一個環節的目標具體化、量化及可控化。
調研工作包括:原研產品的新藥保護期信息、FDA橙皮書中收錄的關於該產品的專利保護內容、原研產品的製劑組成、相關專利提供的可能的製劑製備方法、以及FDA推薦的溶出方法和BE實驗方案等;
檢測工作包括:原研產品的外觀尺寸、藥物含量、雜質及藥物溶出曲線等測定數據。
對原研產品的處方及工藝越了解,則越有利於仿製藥的研發。然而,FDA明確披露的隻有處方中的輔料類別,即產品由原料藥與哪些輔料組合而成。而對於每種輔料所占的確切比例,以及製劑的實際製備工藝並無介紹,通常需要通過一些相關專利或文獻的信息披露推測獲得,這也是仿製藥產品要摸索的最關鍵的環節之一。如果專利中明確保護了某種輔料或某個工藝,那麽在專利過期前需要規避並選擇其他解決方案,這就使仿製藥的製劑研發難度大大提高。
對原研產品理化性質的測定幫助我們更好的理解目標性狀。對於口服固體製劑來說,通常在溶出實驗中具備與原研片一致性較高的產品才有更高的BE實驗通過率。因此在這一階段,我們最關注的是所研發仿製藥的溶出曲線與原研產品的匹配情況,而如何開發體內外相關性高的溶出方法,更是BE實驗獲勝的關鍵。
在實驗室小試階段,通過對處方及工藝的篩選及優化,獲得與原研產品溶出度非常接近的產品,並明確處方中的關鍵輔料及工藝中的關鍵參數對溶出曲線的影響趨勢。值得注意的是,搞清楚影響趨勢,要比“碰運氣”找到了最佳溶出曲線重要得多。
中試放大階段
批量通常在20kg~50kg
優化獲得了與原研產品較為接近的備選處方後,即可進入車間中試放大階段。通常從這一階段開始到未來申報生產及商業化生產等均要求在cGMP環境下操作。
從小試到中試,是批量變化最大的一步,也是最容易產生“放大效應”的一步,處理好放大效應是產品最終走向申報乃至商業化生產的成功關鍵。
放大效應有兩個方麵:一是參數的設計問題,即如何參照小試的設備工藝參數來設定中試放大中大設備的工藝參數。打個比方說,煮1個雞蛋如果需要10分鍾,那麽煮20個雞蛋通常不隻是相同的10分鍾,也一定不需要等比擴大20倍後的200分鍾。因此,推算並確定中試放大的關鍵工藝參數是一個難度較大的問題。
另一個容易出現的放大效應是,有些問題在小試實驗中不存在或很小,到中試生產才顯現出來並存在嚴重缺陷,也就是放大生產可行性的問題。例如,一些產品在實驗室小試中並無壓片問題,但在中試生產高速旋轉壓片時可能出現物料粘衝、分層、可壓性差的問題,這些問題隻有在批量較大時才會顯現,因此即使小試研發的產品質量再好,在未來也是無法量化生產的。
在解決了上述“放大效應”問題後,我們來關注產品質量。大部分產品在研發時,相同的處方及工藝在小試和中試放大分別得到的產品的溶出曲線存在一定差異,通常會返回實驗室進行調查研究,以及重複放大生產進一步優化參數設計。當然,如果在小試研發階段對溶出曲線變化趨勢的影響因素研究的十分清晰透徹,也可更快更準確地使產品接近目標值。
這樣的交叉循環反複工作可能持續半年到兩年的時間,甚至更久,視產品研發難度及放大效應大小而定,最終目標是獲得與原研產品體外各方麵指標一致且質量可控的中試產品。
申報生產階段
批量通常在50kg~150kg
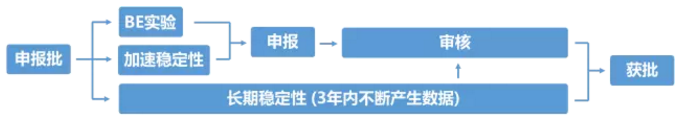
申報生產是整個研發過程中最為關鍵的一個環節,也是申報材料中FDA最關注的生產批次。目前FDA的要求是,須連續生產三批且每批批量不小於10萬片(粒)的產品。這裏的連續,並非中間一天不能停歇,而是中間不能再穿插任何放大生產批次。
由於申報批的批量與中試批量相差不多,為了節省時間及成本可視研發難度等情況考慮是否在申報批前做1-2批與申報批次同等批量的放大研究。
三批申報產品生產完成後,需要同時開展長達6個月的加速穩定性實驗和長達3年的長期穩定性實驗,隻選擇其中一批進行BE實驗(通常是第一批),並撰寫及整理申報材料。6個月加速穩定性實驗結束後,如果檢測結果符合標準,且已通過BE實驗,則這三批申報產品具備申報資格,可以向FDA遞送材料進行申報。材料遞送後,FDA還需要1-2年審批時間,中間會提出一些缺陷性問題,藥企需回答問題,並可能還需要補充小試或中試實驗數據,同時補充不斷獲得的長期穩定性實驗數據。
在申報生產環節,最為關鍵同時也是難度最大的一個步驟是BE實驗。隻有BE實驗成功的申報批產品,才意味著在體內的釋放及吸收行為與原研產品完全一致。美國市場仿製藥BE實驗的成功率僅為48%左右。一旦BE失敗,三批申報產品全部報廢,研究者要對失敗原因進行全麵的調查和評估。
通常情況下,如果BE實驗結果與原研產品相差甚微時,有以下兩種途徑快速重新完成申報工作。①如果僅僅是在生產過程中工藝參數控製不當,可在設置範圍內做細微調整,並立即重新開始三批申報工作;②如果需要調整的工藝參數超出生產文件設置範圍,則需要修改升級生產文件後,再進行申報生產。
如果BE實驗結果與原研產品相差較大時,很可能就要返回到最初的實驗室小試階段重新開始研究。
可能有人會問,體外溶出實驗中申報產品已與原研產品具有較高的溶出匹配度,為什麽體內BE實驗還會失敗?一般來說,未找到更具區分性或與體內相關性更好的溶出方法是BE失敗的主要原因。因此實驗室小試研究需要這可能是因為缺乏深入全麵的體外溶出實驗研究。著重確定最佳的溶出分析方法,並在新方法下重新優化處方及工藝。
結語
製劑出口海外市場前景廣闊,但對製劑質量的要求也極高,所需研發周期較長,成本較高,因此對於藥企來說,需要具備豐富的製劑技術經驗與資本積累,才有可能生存下去。這也極大推動了中國藥企製劑水平的飛速發展,使市場建立起優勝劣汰的健康的競爭模式。




























 相關新聞
相關新聞

 關於我們
關於我們










