利好一:參比製劑備案信息公布,一致性評價趨透明
9月12日,中檢院公布了參比試劑的相關信息,分別是中檢院推薦參比製劑品種信息和截至2016年6月30日企業提交的參比製劑備案信息。這符合CFDA關於落實《國務院辦公廳關於開展仿製藥質量和療效一致性評價的意見》有關事項的公告(2016年第106號)的精神,一致性評價的信息也越來越公開透明。
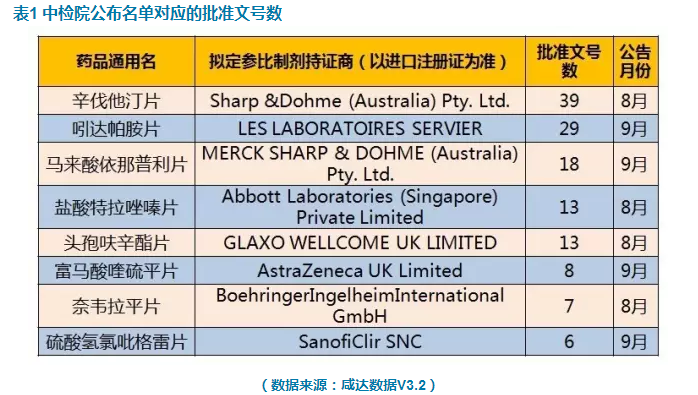
中檢院推薦參比製劑品種信息在2016年8月8日和9月12日各發表4個品種,所公布的8個品種皆是289目錄品種(2018年底前須完成仿製藥質量和療效一致性評價的品種),並且這8個品種的原研參比藥都來自歐洲。
中檢院所公布的8個品種中,批準文號數最多的是辛伐他汀片,其次是吲達帕胺片,第三是馬來酸依那普利片。
企業提交的參比製劑備案信息可以看出哪些產品是企業重點關注的。從中檢院提供的企業參比試劑備案可以看出,截至2016年6月31日,共942個廠家產品參與了備案,其中509個廠家產品在289個仿製藥目錄內。
鹹達數據V3.2發現,本次中檢院942個品種中,阿莫西林膠囊是最多企業備案的產品,其次是辛伐他汀片,第三是苯磺酸氨氯地平片。
以辛伐他汀片為例,比較中檢院和企業備案的情況後可以發現,企業備案和中檢院都鎖定參比試劑的公司為默沙東,但企業備案對原研藥生產廠家的英文名並沒有統一名稱,較難統計。
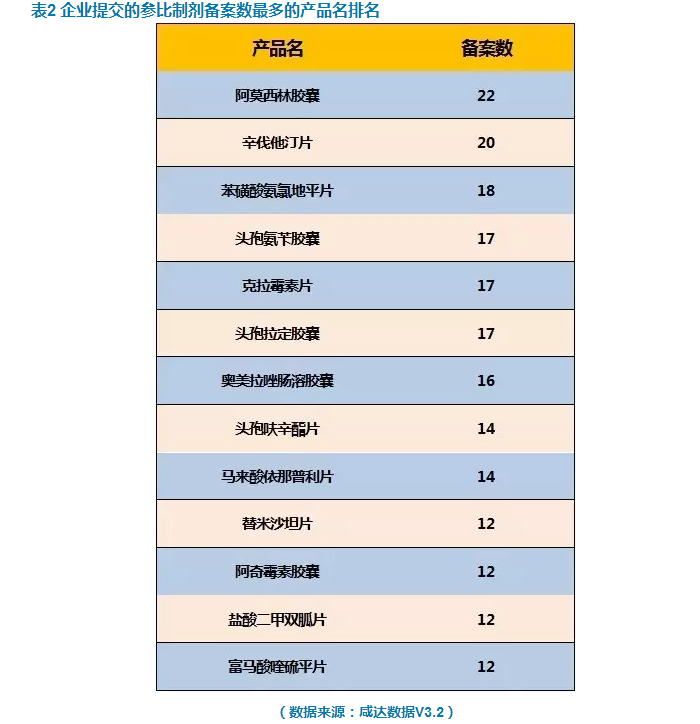
根據中檢院的數據,289目錄產品有138個產品共509個品規已有企業備案原研,這意味著有將近50%的產品通用名目前企業已有初步研究意向。其中,阿莫西林膠囊和苯磺酸氨氯地平片是289目錄中最多備案的產品,各有18個品規已備案。
鹹達數據V3.2將企業備案的藥品數對比CFDA於8月17日所發布的2018年底前須仿製藥質量和療效完成一致性評價品種批準文號數,發現頭孢呋辛酯膠囊劑與片劑、阿卡波糖膠囊、複方利血平氨苯蝶啶片、琥珀酸亞鐵片、環孢素膠囊、阿法骨化醇片、富馬酸比索洛爾片和阿卡波糖片目前企業的備案數已經等同目標完成一致性評價品種批準文號數,其中阿卡波糖膠囊、複方利血平氨苯蝶啶片、琥珀酸亞鐵片和環孢素膠囊,都是隻有1個批準文號需要做試驗的藥品。
942個企業備案數據中,備案最多的是山東新華製藥股份有限公司,其次是石藥集團歐意藥業有限公司,第三是山東京衛製藥有限公司。而2018年必須完成的289目錄品種中,備案最多的企業是山東新華製藥股份有限公司,其次是石藥集團歐意藥業有限公司,第三是華潤雙鶴利民藥業(濟南)有限公司。
小結從數據來看,需要2018年完成一致性評價的基藥口服製劑17,740個標準文號數僅有509個備案,不到3%,這意味著許多廠家還是在觀望。所幸的是,289目錄通用名中有138個藥品有備案記錄,亦即48%個藥品已有廠家有意向觀望,其中在289目錄中隻有1家申報的已備案藥品共有45個,包括批文號數較多的阿司匹林腸溶片和雙氯芬酸鈉腸溶片。
北京近日對289目錄的產品有高達300萬元的資金支持,這將會激勵北京企業對289目錄藥品一致性評價的參與度。
利好二:“改規格藥品評價一般考慮”政策發布
9月13日,CFDA公開征求仿製藥質量和療效一致性評價改規格藥品評價一般考慮的意見。這意味著此前大家一直爭議的改規格藥品的一致性評價該怎麽做有了新的補充。
CFDA對改規格藥品有了新的定義,改規格藥品指該規格在歐盟、美國或日本均未獲準上市或雖獲準上市但無法確定同規格參比製劑的藥品,包括原研藥品相應規格曾獲批但已不存在或原研藥品從未獲批該規格的品種。
值得注意的是,CFDA今年發布的化學藥品新注冊分類中,要求仿製藥必須與原研藥具有相同活性成分、相同劑型、相同給藥途徑、相同規格;對於改良後增加規格的,需按新藥受理。改規格藥品的評價包括藥學和臨床試驗等多個方麵的內容。
改規格藥品需充分論證改規格藥品存在的科學性、合理性和必要性,並且藥品規格的變更應在其臨床使用的用法用量範圍內,亦即其規格應在單次最小給藥劑量與單次最大給藥劑量的範圍內。
藥學研究同樣是改規格藥品重點研究的內容,包括處方組成與工藝研究、質量標準與質量控製,以及增加規格產品的穩定性試驗研究。
試驗要求方麵,滿足以下條件者,可選擇原研同品種其他規格為參比製劑,以相同劑量給藥進行人體生物等效性試驗:仿製藥和原研藥的適應症和用法用量相同;在治療劑量範圍內,藥物呈現線性藥代動力學特征;在治療劑量範圍內,藥物呈現線性藥代動力學特征;改規格製劑與參比製劑的活性組分一致,且製劑處方比例相似;改規格製劑和參比製劑體外溶出、釋放特征相似。不滿足上述條件的改規格藥品,CFDA建議采用臨床試驗進行評價。
小結適用於一致性評價的改規格藥品有了新的指導補充文件,對此類藥品有了新的定義和相關的研究準則,這對於相關企業是利好。
但是,對於改良型改劑型的企業而言,目前還沒有相關的文件發布。預計改劑型藥品的療效再評價的項目還是要做臨床試驗。
利好三:參比製劑找不到?優先選擇安慰劑對照
9月14日,總局辦公廳公開征求仿製藥質量和療效一致性評價臨床有效性試驗一般考慮的意見。這是主要針對找不到參比製劑的仿製藥提供臨床有效性試驗如何進行的補充指導。
對於此類找不到參比製劑且國內已上市的仿製藥,在進行臨床有效性試驗前應回顧並評估該藥品在現有治療中的臨床價值,並基於其國內外臨床研究及其應用情況(如指南的地位,目前循證醫學證據的情況),對該藥品的臨床有效性進行初步判斷。生產廠家應考慮該藥物的臨床療效情況,與其他治療藥物的療效比較情況,以及是否存在影響現有治療藥物療效的其他因素(如耐受性、依從性或患者傾向性)。
為了證明藥品的有效性,CFDA鼓勵盡可能選擇安慰劑對照進行臨床有效性試驗,除了細胞毒類藥品等特殊情況不適合應用安慰劑對照而必須選擇陽性對照藥的情況。
此外,陽性對照藥應適應症相同且最好機理相同,臨床療效確切且可預期,並且是公認的、有良好循證醫學證據。不過,CFDA沒有硬性規定陽性對照試驗,從而相對降低了有效性試驗的難度。
仿製藥質量和療效一致性評價臨床有效性試驗的比較類型主要包括優效性試驗和非劣效性試驗。
優效性試驗的目的是顯示試驗藥的治療效果優於對照藥,非劣效性試驗的目的是確證試驗藥的療效至少不差於陽性對照藥。CFDA鼓勵生產廠家盡可能選擇優效性試驗設計而沒有硬性要求非劣性試驗,從試驗設計上又一次降低了臨床有效性的難度。
CFDA認為,作為已上市仿製藥,其療效和安全性循證醫學證據已有一定積累,臨床終點可以使用有價值的替代終點或生物標記物,以科學、準確、靈敏、有效地實現試驗目的。這意味著此類仿製藥無需像新藥臨床試驗那般嚴格考核臨床終點。
小結:這對找不到參比製劑的且具有臨床療效的仿製藥是個利好,特別是曾在海外上市但現在退市的原研藥及其改劑型的藥品。在臨床從嚴的背景下,雖然臨床試驗的難度有所降低,但是安全無效的藥品仍難以獲得更優的指向性結果。




























 相關新聞
相關新聞

 關於我們
關於我們










